
Research Project 3:
The role of AP-5 in the endolysosomal system, autophagy and autophagic lysosome reformation
Principal Investigator
Prof. Dr. med. Christian Hübner
Postdoctoral Fellow (FOR2625 funded)
Dr. rer. nat. Mukhran Khundadze

Project Summary
Hereditary spastic paraplegia (HSP) is characterized as a progressive spastic gait disorder that arises from a length-dependent dying back degeneration of corticospinal tract fibers. Distinct genetic HSP entities (SPGs) can be distinguished. SPG48 is caused by mutations in AP5Z1. Its gene product was shown to co-precipitate with 5 additional proteins, among them Spatacsin and Spastizin, which are associated with HSP subtypes SPG11 and SPG15. The proteins were later identified as the subunits of a novel adaptor protein complex 5 (AP-5) with the gene product of AP5Z1 as its ζ subunit and Spatacsin and Spastizin as likely components of the scaffold. Recent evidence supports a role of AP-5 as a backup system of retromer to promote the recycling of transmembrane proteins to the trans-Golgi network (TGN) with the cation independent mannose-6-phosphate receptor (CI-MPR) and sortilin as likely cargo receptors. We could show that KO mice for the ζ subunit of AP-5 develop a progressive spastic gait disorder characterized by progressive accumulation of intraneuronal autofluorescent material and axon degeneration. KO mouse embryonic fibroblasts displayed structural alterations of the Golgi and a block in the autophagic flux under stressed conditions. Moreover, the recycling of lysosomes from autolysosomes, a process also known as autophagic lysosome reformation (ALR), was compromised. Still it is an open question, why the disruption of AP-5 results in the altered Golgi structure, accumulation of some Golgi-related proteins in lysosomes, impaired autophagic flux at stressed conditions and compromised ALR. Our preliminary data from the first funding period suggest that the Pi4k2a abundance and distribution are altered in MEFs and in Purkinje cells of Spg48 KO mice. Because Pi4k2a is a key enzyme for the production of PI(4)P, which is involved in a variety of membrane trafficking and signaling events including exocytosis and autophagy, we hypothesize that alterations of PI(4)P-related phospholipid signaling could be the common basis of the different defects. Therefore, we will compare the subcellular distribution of Pi4k2a and its product PI(4)P upon disruption of AP-5. We will further study how the overexpression, knockdown or knockout of Pi4k2a affects autophagy, lysosome function and ALR. In addition, we will investigate whether the knockdown or the pharmacological inhibition of Pi4k2a can restore defects observed upon disruption of AP-5. To understand how the redistribution of Pi4k2a relates to AP-5, it will be essential to localize AP-5 in living cells. Therefore, we will generate cell clones with a fluorescently tagged ζ-subunit of AP-5 and visualize AP-5 at steady state and upon induction of autophagy and ALR. To get further clues about possible cargo receptors we will perform pull-down assays and a TurboID approach to determine proteins interacting with the ζ-subunit of AP-5 at baseline and upon starvation.
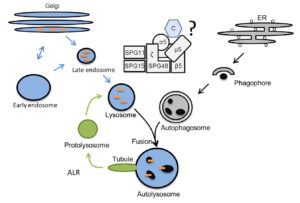
What is the role of the AP-5, SPG11, SPG15 complex within the degradataive pathway?The Golgi and the endolysosomal pathway are labeled in blue, autophagy in grey, and ALR in green. Orange dashes: lysosomal enzymes, Black circles: autophagic material. ER: Endoplasmatic Reticulum, C: cargo
References
Khundadze M, Ribaudo F, Hussain A, Rosentreter J, Nietzsche S, Thelen M, Winter D, Hoffmann B, Afzal MA, Hermann T, de Heus C, Piskor EM, Kosan C, Franzka P, von Kleist L, Stauber T, Klumperman J, Damme M, Proikas-Cezanne T, Hübner CA. A mouse model for SPG48 reveals a block of autophagic flux upon disruption of adaptor protein complex five. Neurobiol Dis 2019; 127:419-31.
Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, Koch N, Hennings JC, Franzka P, Huebner AK, Kessels MM, Biskup C, Jentsch TJ, Qualmann B, Braulke T, Kurth I, Beetz C, Hübner CA. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet 2015; 11:e1005454.
Khundadze M, Kollmann K, Koch N, Biskup C, Nietzsche S, Zimmer G, Hennings JC, Huebner AK, Symmank J, Jahic A, Ilina EI, Karle K, Schöls L, Kessels M, Braulke T, Qualmann B, Kurth I, Beetz C, Hübner CA. A Hereditary Spastic Paraplegia Mouse Model Supports a Role of ZFYVE26/SPASTIZIN for the Endolysosomal System. PLoS Genet 2013; 9:e1003988.